What is Fastachar for and how to use it?¶
Fastachar is a graphical user interface to the fastachar python module that allows a user to compare pre-aligned DNA sequences. Sequences of different markers must be analysed individually (not concatenated). A typical application is to distinguish one species from a set of different, but closely related species, based on DNA sequences.
Example¶
Let’s assume we have DNA sequences from specimens of cryptic species (a pair or more). After the discovery of the new species it is paramount to carry out the final step in taxonomy, their description. However, in this case, the morphological characters per se cannot be used to describe the new cryptic species. Therefore, molecular diagnostic characters (present in all members of a taxon and absent in all other taxa) can be obtained from the DNA sequences. These characters can be used to describe a species in a similar fashion to traditional morphological diagnostic characters used for species descriptions. To that end, the algorithm compares two sets of sequences, with one set consisting of a number of sequences of a taxon (e.g., a new species) and the other consisting of sequences of other taxa (e.g., congeneric or confamilial species). For each homologous position in the alignment (pre-aligned sequences), the algorithm tests for all characters of the sequence in the first set to be the same and to be different from all other characters of the sequences in the other set. When these conditions are met, the character in that position (nucleotide or amino acid) is marked as a molecular diagnostic character.
Preparation¶
The input for fastachar is a list of DNA sequences, formatted in the fasta format (see also https://en.wikipedia.org/wiki/FASTA_format). The program assumes that the DNA sequences that are going to be compared already:
- are aligned, and
- written into a single file in fasta format.
There are several software programs to align sequences (e.g. Mega and Geneious).
Running Fastachar¶
On Windows, fastachar is run by executing the fastachar.exe, and on Linux, it is run by executing fastachar from the terminal console. Once started, a new window appears with three empty text boxes, labelled “Unselected species”, “Selected species list A” and “Selected species list B”, respectively. Below, there is a set of radio buttons to select the comparison operation, a button to execute the comparison (“Process”) and a button (“Clear output”) to clear the output that is generated and shown in the bottom text box, see the Figure.
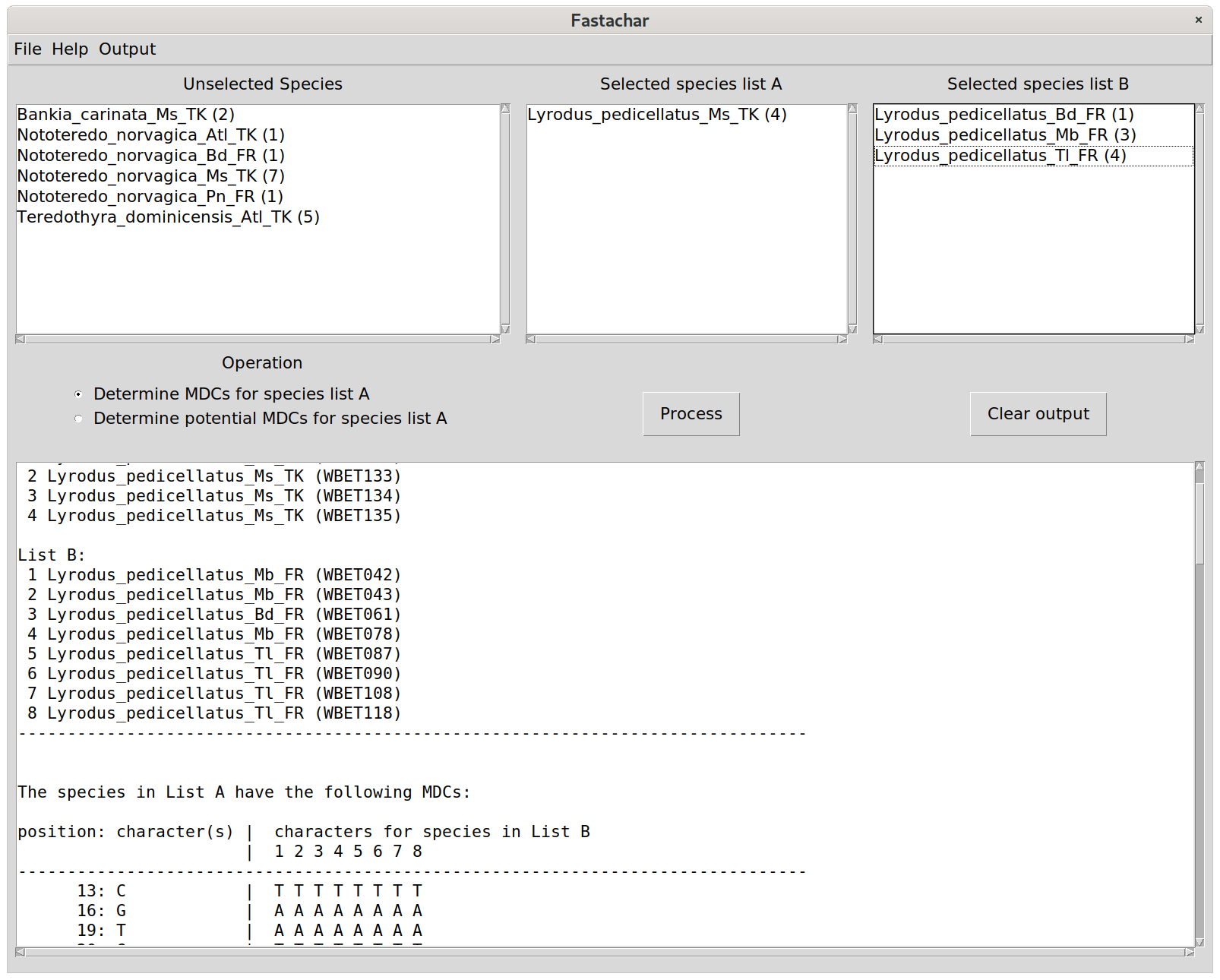
Figure 1: Main window of Fastachar
Opening a fasta file¶
To start working, a fasta file is opened using:
File
└── Open fasta file
and select a fasta file from the dialogue offered. If a valid fasta file is read, the text box Unselected species is populated with the names of the species found.
Alternatively, a fasta file can be opened using:
File
└── Open fasta file /w preview
This allows the user to specify how the fastachar should interpret the header strings that precedes each string of sequence data. When opening a file with preview, first a pop-up window appears, similar to the one below
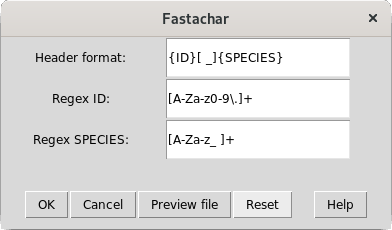
Figure 2: A pop-up window allowing to specify regular expressions for parsing the fasta header strings
This window provides the user with a way to specify how the header strings are to be parsed. All three fields accept regular expressions, see Regular expressions in FastaChar for more information on regular expressions and a worked example.
- Header format::
- The Header format describes how each header is structured and must contain the strings {ID} and {SPECIES}. In the example given, the id precedes the species name and a space or an underscore separates the two strings.
- Regex ID::
- The value for the entry Regex ID is substituted for the string {ID} in the header format string. As this string should match any of the lab codes or IDs used in the fasta file headers, it will usually be a regular expresssion.
- Regex SPECIES::
- The value for the entry Regex SPECIES is substituted for the string {SPECIES} in the header format string. Also this string will usually be a regular expresssion.
After editing the regular expressions, the button Cancel cancels the modification, whereas the button OK accepts them. The button Preview file provides the user with a file chooser dialogue to select a fasta file. After this selection, the file is opened, and parsed. Each header is interpreted and how it fares is shown in a separate window:
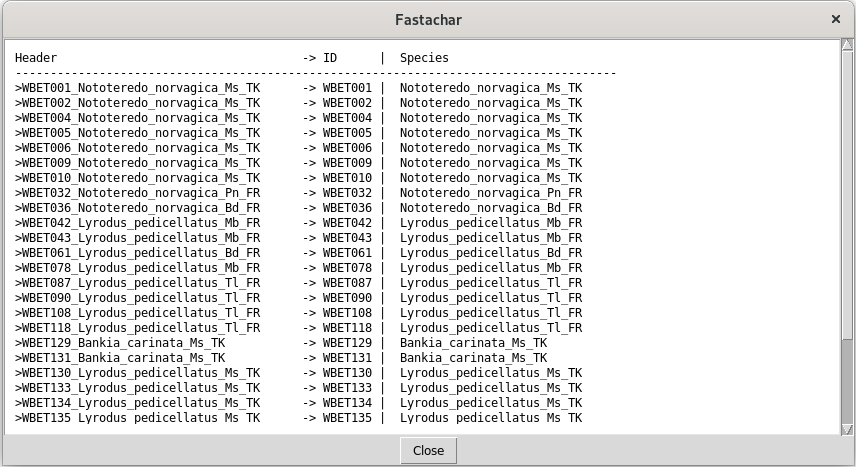
Figure 3: Popup window showing the results of the header parsing.
In the example of Figure 3, we see in the left column (Header) the string as it appears in the fasta file. In the middle column, the parsed ID string is shown, and in the right column the species name. If the parsing fails completely, dashes only are shown. If the regular expressions do not match the format of the header strings, erroneous results are displayed.
Note
If some how the program is not capable of parsing the strings correctly, a work around would be to describe the header string as {SPECIES}{ID}, leave the regex for the ID blank, and for the SPECIES a regular expression .+ is prescribed. FastaChar will now ignore any id’s and consider the header of each sequence as a separate species.
Selecting species for lists A and B¶
Select a species name by left-clicking. A multiple selection can be made by clicking again with ctrl pressed, which also selects the item clicked. If instead of ctrl the shift key is pressed, all the items in between are selected as well.
In order to move them into either list A or list B, drag the selected items from the Unselected species text box to the target text box whilst holding the right-mouse button pressed.
Selecting the operation¶
Once the selection is made, the comparison operation is to be selected. Two operations are implemented:
- Determining MDCs for species list A
- Determining potential MDCs for species list A
After selecting the operation, the operation is executed by clicking the Process button, and a report appears in the lower text box, see Figure 1.
The output lists the path of the input fasta file (not shown in Figure 1), the species’ names and IDs of the sequences in list A, and list B. If the species in list A have any molecular diagnostic characters, then they are listed by their position, their value, and the values of the sequences in list B for the same position. Note that masked characters, if any, are left blank.
A molecular diagnostic character is the character at position \(k\) of the sequences in list A, for which holds that:
- all characters in A are identical for this position, and
- all characters in B for are different from those in A for this position.
For a potential diagnostic character, the second condition is met only. For a precise definition, the user is referred to the accompanying paper, see [Merckelbach2020].
Case files¶
To facilitate repeated operations on a specific file, or storing the specifics of a case for future reference, a so-called case file can be used. When writing a case file via:
File
└── Save case file
the following information can be stored:
- the fasta file read
- the regular expressions used for reading
- the selection made
- the operation selected.
A previously saved case file can then be loaded by
File
└── load case file
Output¶
Multiple operations as well as species selections can be processed and the output will be appended to the lowest text box. The output can be cleared using the Clear output button.
To save the output to file, select from the menu:
Output
└── Save report (txt)
to write the output of the last operation as shown in a text file, or
Output
└── Save report (xls)
to write the output in an excel file, with a tab for each processing operation.
Help¶
The user interface also provides help and information on the licensing from the menu entry:
Output
└── Help
and
Output
└── About
respectively.